LA SINDROME DI ALGELMAN L’IMPORTANZA DEI PERCORSI RIABILITATIVI
Ebook :I COMPORTAMENTI PROBLEMA NELLA SINDROME DI ALGELMAN di Sara Nappi per Ass. La Nostra Famiglia
https://www.medicinaxtutti.it/2020/05/03/2289/ post n. 38

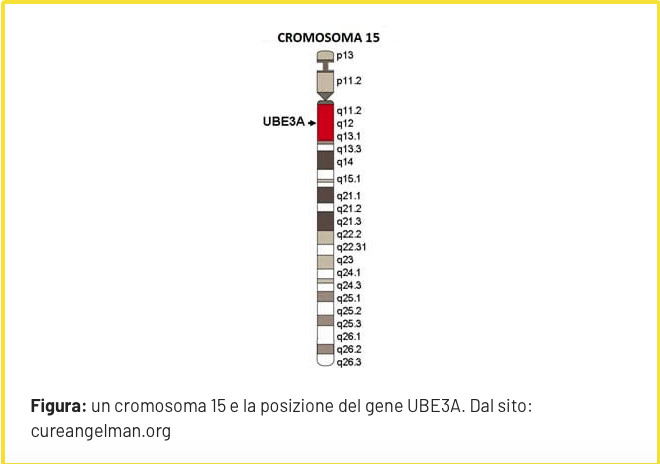
La Sindrome di Angelman (SA) è una sindrome genetica, fu descritta per la prima volta nel 1965 dal pediatra inglese Harry Angelman, da cui prende il nome. In questa sindrome manca una porzione, contrassegnata come 15q11-q13 (UBE3A), del cromosoma di origine materna numero 15. A causa di un complesso meccanismo biologico, l’imprinting, i geni contenuti in questa porzione di cromosoma sono funzionanti solo nel cromosoma materno, e sono invece silenti in quello paterno.
Epidemiologia e sintomatologia
La sindrome di Angelman è piuttosto rara colpisce un soggetto su 20000. In Italia sono circa 6.000 i puppet children, cioè i bambini colpiti da questa malattia che vengono così definiti per i caratteristici movimenti a scatto, lo sguardo terso, il sorriso facile, la postura “a marionetta”. Il rischio di sindrome di Angelman nei fratelli varia tra 1% e il 50% . La consulenza genetica è indicata per i familiari di primo grado del soggetto con SA, e per i per familiari più lontani.
I bambini affetti nascono apparentemente sani ma dopo la nascita si manifestano nel neonato disturbi alimentari quali difficoltà di suzione e reflusso gastro-esofageo, a questi sintomi si associano scarsa mobilità o “mollezza” muscolare per ipotonia muscolare generalizzata e pianto anomalo. Intorno ad un mese di età compaiono altre evidenze cliniche: il sorriso frequente anche in assenza di stimoli e gli accessi di riso parossistico, che spesso portano ad attacchi epilettici e crisi notturne repentine. Vi è un ritardo del neurosviluppo, i bambini Angelman presentano inoltre ritardata o diminuita crescita della circonferenza cranica, che generalmente comporta la presenza di microcefalia (cranio piccolo) intorno ai 2 anni. È presente epilessia che compare prima dei 3 anni di vita associata ad anomalie elettroencefalografiche.
Il quadro clinico è caratterizzato dalla presenza di una grave disabilità intellettiva, linguaggio è gravemente compromesso o assente mentre viene in parte conservata la comunicazione non-verbale e vi è deficit dell’attenzione. Si hanno disturbi neurologici che riguardano l’ andatura con deambulazione a base allargata e mancata coordinazione muscolare che rende difficoltosi i movimenti volontari (atassia). Vi sono movimenti stereotipati delle mani; iperattività dei riflessi degli arti inferiori. Inoltre i puppet children, che sono generalmente di pelle chiara con capelli e colore degli occhi chiari, hanno un comportamento tipico caratterizzato da apparente stato di contentezza, crisi di riso, eccitabilità ed iperattività senza aggressività. Infine questi bambini soffrono di anomalie del ciclo sonno-veglia.
Diagnosi
Spesso la diagnosi di Sindrome di Angelman non viene fatta tempestivamente poiché all’inizio i sintomi presenti nel neonato non sono specifici e possono essere sottovalutati. Solo quando, con la crescita del bambino, i sintomi peggiorano viene posta una diagnosi adeguata. Inoltre esiste un basso livello di informazione di pediatri e genitori e degli operatori quali educatori di asili nido/scuola materna, psicologi, fisioterapisti, logopedisti che non facilita il riconoscimento precoce della SA. Il sospetto clinico viene dalla coesistenza dei sintomi ma occorre la diagnosi molecolare per confermarlo. Attualmente sono utilizzati diversi metodi che permettano di rilevare l’alterata espressione o la perdita di funzione della copia del gene UBE3A ereditato per via materna:
- l’ibridazione in situ fluorescente (FISH) è un test che mostra se mancano delle porzioni di cromosoma.
- la metilazione del DNA è una procedura complessa co cui si constata se il cromosoma 15 materno (contenente uno dei due alleli del gene UBE3A) è mancante oppure no. È considerato negativo se il cromosoma 15 materno è presente.
- la ricerca di mutazioni genetiche è un test alternativo alla metilazione del DNA, quando questa dà esito negativo (ovvero il gene UBE3A c’è), ma tutti i sintomi sono, comunque, a sostegno della sindrome di Angelman. Il gene UBE3A, infatti, può essere presente, ma mutato.
Terapia riabilitativa
La Sindrome di Angelman non porta alla morte chi ne è affetto, ma comporta delle disabilità che richiedono un percorso riabilitativo continuo, per garantire ai malati una vita decorosa e un minimo di autonomia.
Uno dei problemi principali dei bambini affetti da SA è la comunicazione per migliorala si ricorre a l’intervento psicoeducativo che ha una matrice cognitivo-comportamentale e si avvale:
- dei principi dell’Educazione Strutturata,
- dell’approccio T.E.A.C.C.H.(Treatment and Education of Autistic and Related Comunication Handicapped Children)
- della Comunicazione Aumentativa Alternativa (C.A.A.).
L’educazione strutturata è un sistema per organizzare lo spazio e il materiale in modo da promuovere l’indipendenza degli studenti. Si utilizzano programmi, strutture, sistemi di studio- lavoro che facilitino l’apprendimento dei compiti. L’educazione strutturata nasce all’interno dell’approccio TEACCH (Trattamento ed Educazione di bambini con Autismo e Disturbi Correlati della Comunicazione), quest’approccio attivo nel North Carolina dal 1972 è un sistema di interventi che comprende attività di ricerca e formazione oltre ad un’organizzazione di servizi che prevede interventi lungo tutto il corso della vita delle persone colpite da Autismo e più in generale da Disturbi Generalizzati dello Sviluppo.
Si propone di:
- modificare l’ambiente in funzione delle esigenze individuali.
- sviluppare al massimo grado le autonomie del soggetto in difficoltà tramite uno specifico programma individualizzato basato sui punti di forza e sulle abilità emergenti di questi individui.
- migliorare la qualità di vita del bambino e dei suoi familiari.
Le modalità di relazione che si usano con un bambino Angelman sono le stesse che si adoperano nell’approccio con qualsiasi altro bambino. I bambini con SA sono empatici, una loro caratteristica è la capacità di riconoscere ed entrare in contatto con lo stato d’animo dell’altro. È partendo dall’empatia che è possibile aprire la strada ad relazione terapeutica efficace. E’ importante sfruttare tutti i canali comunicativi. Per questo occorre utilizzare ciò che visivamente può compensare le difficoltà dovute all’assenza di linguaggio: la mimica facciale, i gesti, le foto, le immagini, i simboli ed i supporti audiovisivi.
La Comunicazione Aumentativa Alternativa non è una tecnica riabilitativa, ma un percorso che rafforza e valorizza le funzioni presenti nel bambino, per migliorare gli scambi comunicativi. E’ detta Aumentativa perché accresce e amplifica le condizioni comunicative già possedute, ed Alternativa perché fornisce un differente sistema di trasmissione dei messaggi alternativo a quello verbale. In pratica vengono utilizzati simboli, immagini , gesti, vocalizzazioni e scrittura. Inoltre la CAA mette a disposizione del malato ausili, tecniche, tecnologie e strategie particolari per compensare le difficoltà comunicative. E’ importante quando si utilizza la CAA il lavoro d’equipe che vede coinvolti oltre agli i operatori C.A.A., i genitori e gli insegnanti. Infatti gli operatori, attraverso il confronto con le persone maggiormente significative per il bambino e attraverso l’osservazione del piccolo nel suo contesto di vita, costruiscono un intervento su misura.
Per intervenire sugli aspetti relazionali, emotivi e di integrazione sociale della SA, è utile la Terapia Multisistemica in Acqua (TMA) che ha l’obiettivo di inserirsi in un progetto riabilitativo globale che considera la specificità del singolo e pianifica un intervento individualizzato e interpersonale. L’acqua è utilizzata come attivatore emotivo e relazionale poiché spinge il bambino a creare un contatto col terapeuta. Quando un bambino è in acqua , infatti, istintivamente si aggrappa all’operatore creando una relazione di vicinanza e affidandosi, sarà poi compito del terapeuta trasformare questa vicinanza istintiva in uno scambio relazionale significativo. Per quanto riguarda la difficoltà comunicativa dei puppet children , attraverso la TMA si cerca di trovare un linguaggio comune fatto di sguardi, di interazione vis a vis e di condivisione di stati emotivi, che portano ad un aumento della reciprocità sociale. Uno dei principali obiettivi terapeutici della TMA , riguarda lo sviluppo di una funzionale regolazione emotiva. La Terapia Multisistemica lavora sul riconoscimento dell’emozione provata, delle motivazioni alla base e sulla risposta più adeguata da fornire al bambino per aiutarlo nella gestione del proprio mondo interiore. Lavorando su questi aspetti si interviene anche sul sistema comportamentale, per attivare una serie di condotte più efficienti e adeguate al contesto e ridurre i comportamenti ritenuti disfunzionali. L’iperattività motoria, l’ipereccitabilità ed i deficit di attenzione sono caratteristiche della Sindrome di Angelman, uno degli obiettivi del percorso terapeutico in acqua è quello di lavorare sul mantenimento del compito richiesto, motivando i bambini con giochi e gratificazioni. Il gioco e l’ambiente acquatico sono motivanti e rendono più gratificante e facile l’accesso all’apprendimento di nuove competenze. Attraverso il gioco, il terapista aiuta il bambino a canalizzare la propria energia ed iperattività così il bambino impara a entrare in relazione con gli altri ed ad interagire con il mondo. L’ambiente acquatico risulta essere utile inoltre da un punto di vista percettivo e motorio, poiché la percezione del corpo in acqua cambia rispetto a come il corpo viene vissuto nella quotidianità.
I deficit del sistema nervoso che si manifestano a livello psico-motorio e intellettivo rendono l’ inserimento dei malati nella società difficoltoso ma la ricerca non si arresta e la speranza di migliorare la vita dei pazienti colpiti dalla Sindrome di Algelman si fa sempre più concreta. Nel 2020 La FAST, in collaborazione con GeneTx Biotherapeutics, ha ottenuto l’autorizzazione dalla FDA – Food and Drug Administration – ad avviare uno studio clinico su GTX-102, ovvero la prima terapia genetica potenzialmente modificante per la SA.
Bibliografia
Adams D et al. “Age related change in social behavior in children with Angelman syndrome” Am J Med Genet 2011;PartA155:1290-1297
Angela MM et al. “Angelaman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes” Trend in Neurosciences 2011;34(6): 293-303
Bird LM et al. “A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome” Am J Med Genet 2011;155:2956-2963
CAA https://www.ilsorrisoangelman.it/la-c-a-a/
Chamberlain ST et al. “Induced pluripotent stem cell models of the genomic imprinnitig disorders Angelman and Prader-Willi syndrome” PNAS 2010;107(41).17668-17673
Conant KD et al. “Epilepsy and the sleep-wake patterns found in Angelman syndrome” Epilepsia 2009;50(11):2497-2500
Daily J et al. “Spatial and temporal silencing of the human maternal UBE3A gene” European Journal of pediatric neurology 2012; 30:1-5
Fiumara A et al. “Epilepsy in patients with Angelman syndrome” Italian Journal of pediatrics 2010;36:31
Grasca CB et al. “Adaptive behavior in Angelman syndrome: its profile and relationship to age” Journal of Intellectual Disability Research 2010;54(11):1024-1029
Harting I et al. “Abnormal myelination in Angelman syndrome” European Journal of Pediatric Neurology 2009;13:271-276
Love V et al. “Sibling relationships in individuals with Angelman syndrome: a comparative study” Developmental Neurorehabilitation 2012;15(2):84-90
Peters SA et al. “Longitudinal follow-up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion class” Journal of Child Psychology and Psychiatry 2012;53(2):152-159
Goldman SE et al. “Sleep in children and adolescents with Angelman syndrome: association with parent sleep and strees” Journal of Intellectual Disability Research 2012;56(6):600-608
Radstaake M et a. “Functional analysys and functional communication training in individuals with Angelman syndrome” Developmental Neurorehabilitation 2012;15(2):91-104
Williams CA et al. “Clinical and genetic aspects of Angelman syndrome” Genet Med 2010:12(7):385-395
Wulffaert J et al. “Maternal parent stress in families with a child with Angelman syndrome or prader-willi syndrome” Journal of Intellectual and Developmental Disability 2010;35(3): 165-174